# 热门搜索 #
大模型
人工智能
openai
融资
chatGPT
十亿参数单细胞基础模型scLong不再只看少数高表达基因,而是把一个细胞里接近 2.8 万个基因 都纳入建模,并结合 Gene Ontology(GO) 的生物学知识,去理解更完整的基因上下文。
在单细胞转录组学领域,研究者希望从每个细胞的基因表达中读出细胞状态、调控关系,甚至预测当某个基因被敲除、某种药物被加入后,细胞会发生什么变化。
过去几年,foundation model(基础模型)开始进入这一领域,显示出强大的迁移能力;但长期以来,现有方法往往为了节省计算,只关注少量高表达基因,忽略了大量低表达甚至零表达基因,同时也缺少对外部基因功能知识的系统整合。这不仅会丢失重要调控信号,也容易让模型对复杂生物过程「只见树木,不见森林」。
近日,MBZUAI、加州大学圣地亚哥分校(UC San Diego)等机构联合团队在 Nature Communications 发表研究成果scLong。

论文链接:https://www.nature.com/articles/s41467-026-69102-y
这是一种拥有10亿参数的单细胞基础模型,基于约4800万个细胞进行预训练,能够在整个人类转录组范围内对约27874个基因建模,并将GO(Gene Ontology) 提供的结构化生物学知识融入模型中。
论文报告显示,scLong在遗传扰动预测、化学扰动预测、癌症药物反应预测、基因调控网络推断等多项任务上,均优于现有单细胞基础模型和多种任务专用模型。
为什么单细胞领域需要一个「更长」的模型?
因为一个细胞并不是只由少数几个「明星基因」决定的。很多现有模型只在约 1500 到 2000 个高表达基因上做 self-attention,这样确实更省算力,但代价是:大量低表达基因被排除在外。
而这些低表达基因虽然「声音不大」,却常常扮演调控开关、信号微调器,甚至在稀有细胞类型、应激反应、疾病进展中发挥关键作用。
换句话说,过去很多模型更像是在读「摘要」,而不是在读「全文」。
另一个问题是,单靠表达矩阵本身,模型未必能真正理解「这个基因是干什么的」。
而Gene Ontology恰恰提供了基因在生物过程(Biological Process)、分子功能(Molecular Function)、细胞组分(Cellular Component)上的结构化知识。过去很多模型主要从数据里「自己悟」,但没有显式利用这些成熟的生物学先验,因此在理解功能关联、调控关系和跨条件泛化时仍然受限。
于是,scLong想做的事情很直接:不仅把基因看全,还要把基因「看懂」。
把一个细胞,读成一整句话
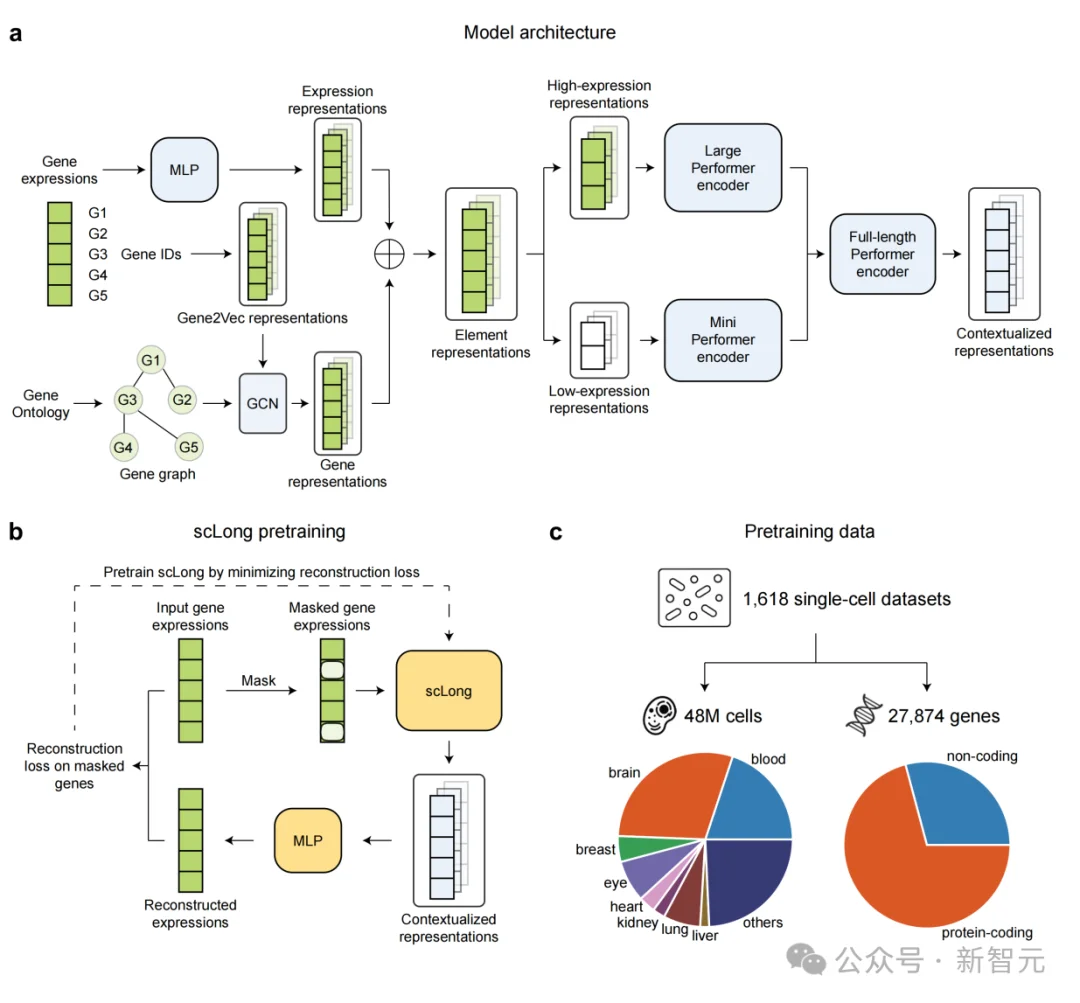
如果用自然语言来打比方,scLong的核心思想很有画面感:把一个细胞的整条基因表达谱,当成一句非常长、非常复杂的话来读。
在这个「句子」里,每个「词」不是普通单词,而是一个 「基因 ID + 表达值」 的组合。模型先用一个表达编码器,把数值型表达量映射成向量;再用一个基因编码器,为每个基因生成带有生物学含义的表示;两者相加后,就得到这个「词」的初始表示。
随后,模型通过上下文编码器,让这些基因彼此「看见对方」,从而学习基因之间在当前细胞中的上下文关系。
这里最有意思的一点是:scLong并没有粗暴地把低表达基因扔掉。 它采用了一个双编码器设计:对高表达基因使用更大的Performer编码器,对低表达基因使用更小的Performer编码器,最后再通过一个full-length Performer把全体基因整合起来。这样既尽量保住了全基因组范围的上下文信息,又在计算量和建模能力之间做了平衡。
更进一步,scLong还把GO知识图谱 接进来了。研究团队先根据基因共享的GO注释来构建基因图:
如果两个基因在生物过程、分子功能或细胞定位上足够相似,它们就会被连接起来;
然后再用图卷积网络(GCN)来学习基因表示。
这样一来,模型不仅知道「这个基因在这个细胞里表达了多少」,还知道「这个基因通常和哪些功能、哪些基因有关系」。这相当于给每个「词」都加了一层背景知识。
预训练方面,scLong使用的是一种类似BERT的思路:随机遮掉一部分表达值,让模型去重建它们。
研究团队用来自1618个单细胞数据集、覆盖50多种组织 的约4800万个人类细胞 进行预训练,覆盖27874个基因,其中既包括蛋白编码基因,也包括非编码基因。对单细胞领域来说,这相当于让模型先在海量真实细胞中「通读语料」,再去做各种下游任务。
还有一个非常值得注意的设计:scLong甚至把零表达也当作信息来建模。 因为零不一定意味着「没意义」,它可能代表「表达太低没测到」,也可能代表「这个基因在该细胞里确实被关闭了」。
前者可能对应弱但真实的生物信号,后者则可能恰恰揭示了某种细胞身份或调控状态。对于单细胞数据来说,这种「把缺席也当作信息」的思路非常重要。
遗传扰动预测:没见过的扰动,也更会猜
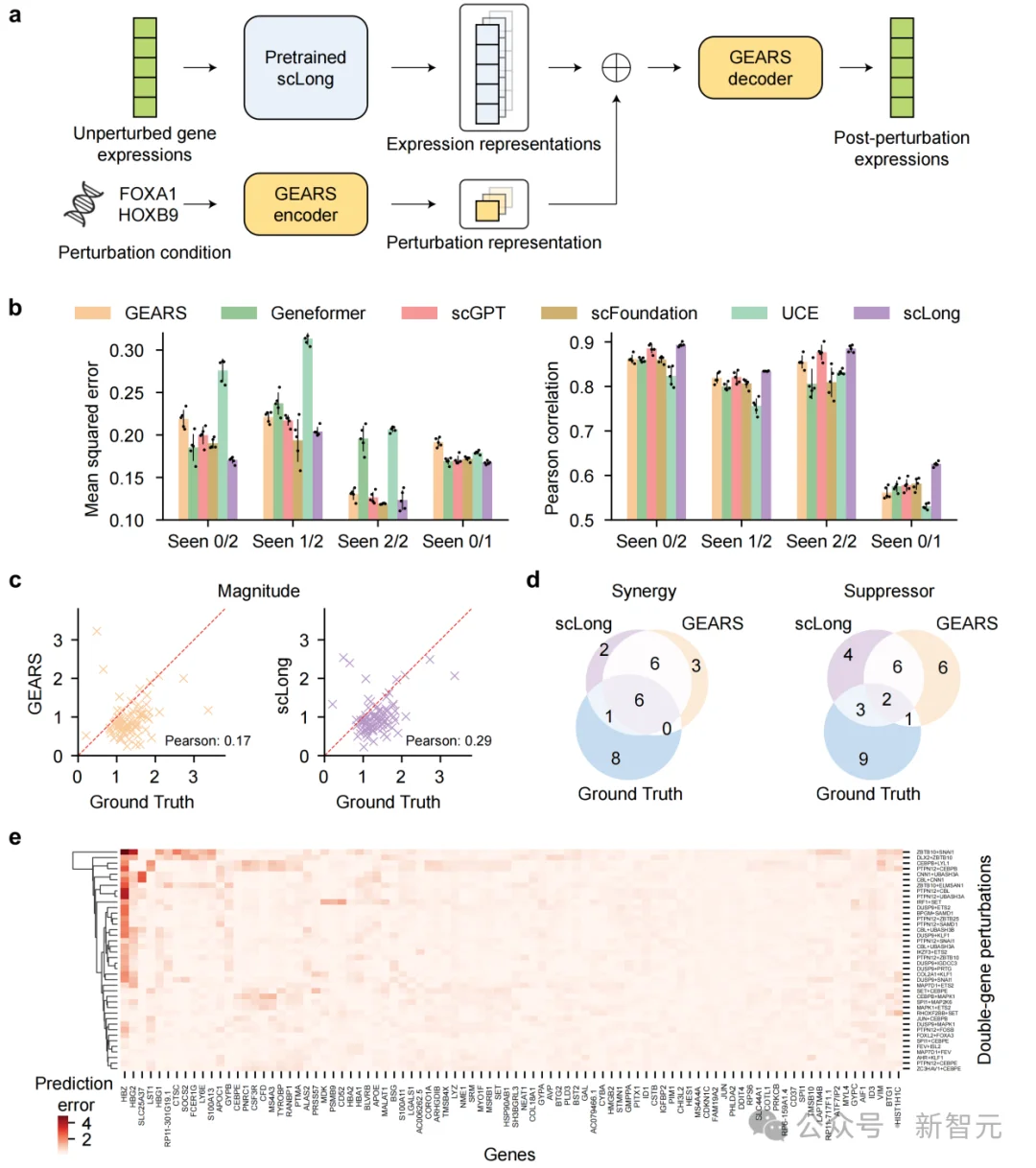
在遗传扰动任务中,模型需要根据细胞扰动前的表达和扰动条件,预测扰动后的表达变化。
论文使用Norman数据集进行评测,并特别关注模型对未见过扰动组合的泛化能力。结果显示,scLong在大多数场景下都优于 Geneformer、scGPT、scFoundation、UCE,以及任务专用模型GEARS、ALM和简单基线No-Change。尤其是在更困难的Seen 0/1和Seen 0/2场景中,scLong的优势更明显:例如在Seen 0/1 场景下,scLong的Pearson相关系数达到0.625,高于GEARS的0.561;在Seen 0/2场景下,scLong的MSE为0.170,也优于多数基线。
不仅如此,scLong对双基因扰动中的协同(synergy)和抑制(suppressor)两类遗传互作的识别也优于GEARS。
这意味着它不仅能预测「会变多少」,还更接近理解「这些基因之间是怎样一起起作用的」。
化学扰动预测:新药上来,先让模型「测一测」
在化学扰动任务中,模型输入药物分子图、剂量和细胞系信息,输出扰动后的基因表达。论文在L1000子集上评估了scLong,结果显示:无论是RMSE、Spearman/Pearson 相关,还是Top-100精度指标,scLong都显著优于Geneformer、scGPT、scFoundation、UCE和任务专用模型DeepCE。
换句话说,面对一个新化合物,scLong更擅长预判它会把细胞「推向什么状态」。
癌症药物反应预测:更懂癌细胞,也更懂联合用药
在癌症药物反应预测任务中,模型需要根据药物结构和癌细胞表达谱,预测药物疗效。论文在DeepCDR数据集上报告,scLong的Pearson相关系数达到0.878,高于Geneformer 的0.852、scFoundation的0.867、DeepCDR的0.837以及线性模型的0.746
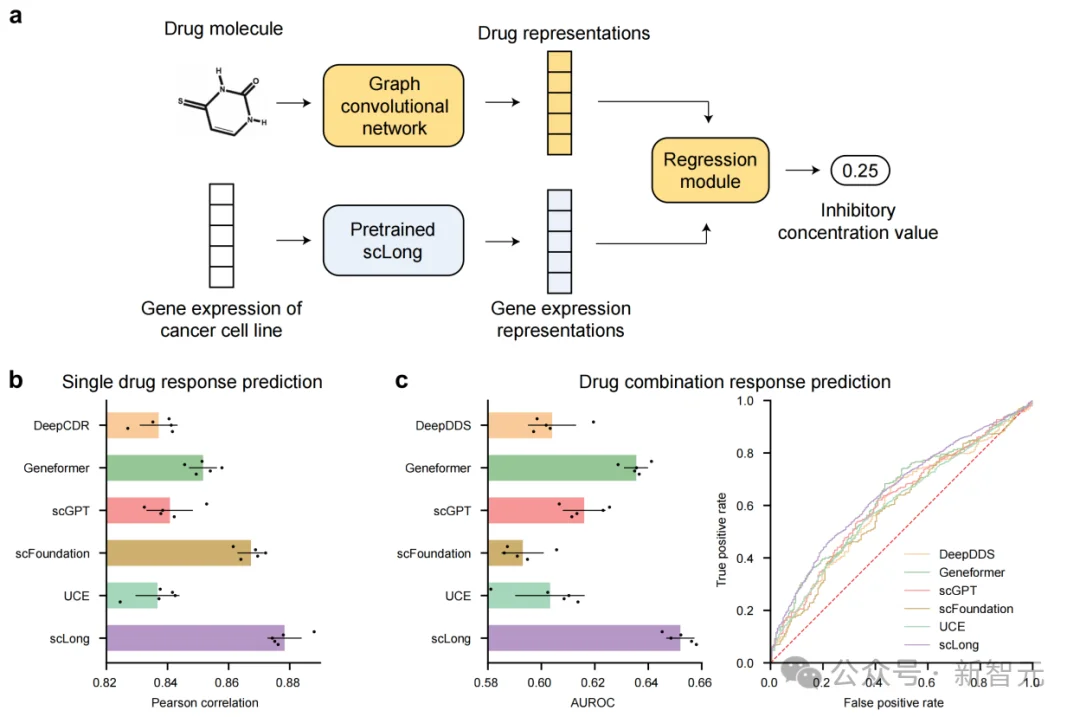
更有意思的是,研究团队还把问题升级到药物组合预测:同一个癌细胞系面对两种药物联用,会不会有更好的反应?
在分布外测试集上,scLong的AUROC达到0.652,同样超过了多种基础模型和任务模型。这说明它不仅能看单药,还能在更复杂的联合治疗场景中提供线索。
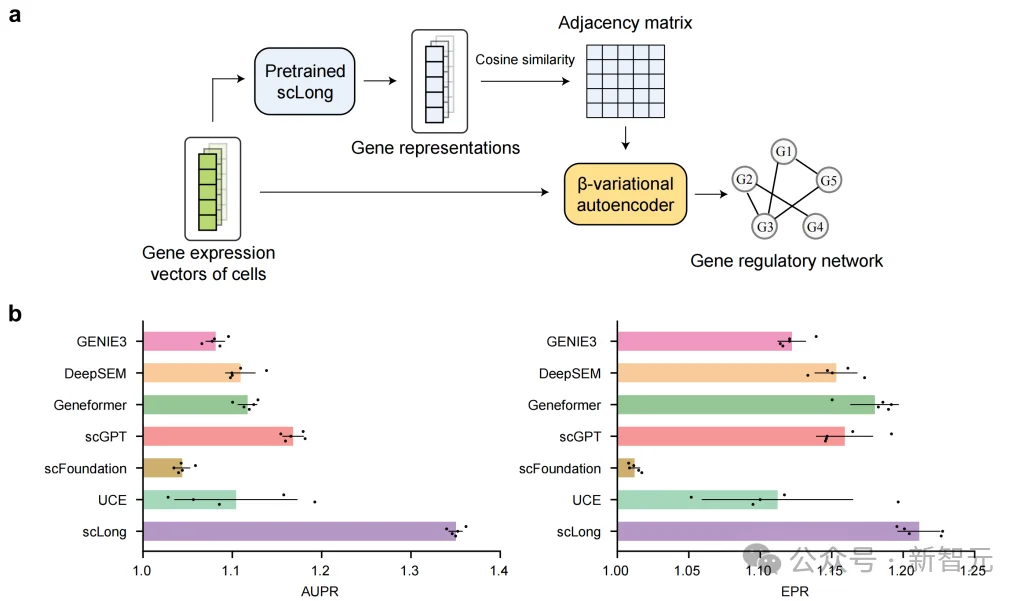
基因调控网络与批次整合:不仅会预测,还会「组织知识」
在基因调控网络(GRN)推断任务中,scLong从基因表示之间的相似性出发,去重建谁调控谁。
结果显示,其AUPR达到1.35,显著优于Geneformer、scGPT、scFoundation、UCE、DeepSEM、GENIE3以及直接使用GO图的基线。
也就是说,scLong学到的并不是「死记硬背」的GO网络,而是结合具体细胞数据后更贴近真实生物系统的关系图。
在零样本批次整合任务中,scLong在pancreas数据集上取得0.96的batch ASW,超过Raw、HVG、scVI以及其他foundation model
值得注意的是,scLong既没有在这个数据集上预训练,也没有微调,却仍然超过了专门在该数据集上训练的scVI,显示出很强的迁移性。
最后,消融实验也给出了很强的支撑:去掉低表达基因建模、或者去掉 GO 图后,性能都会下降。这说明scLong的提升不是偶然,而正是来自「看全基因」和「引入生物知识」这两件事本身。
从「看少数基因」走向「看全基因组」:它把约 2.8 万个基因都纳入上下文建模,而不是只盯着高表达基因。
把生物知识真正嵌进模型:GO 不再只是注释表,而是参与到基因表示学习的核心结构中。
大规模预训练带来强迁移能力:基于 4800 万细胞的预训练,让模型在多个下游任务上都能稳健发挥。
不只是「更大」,而是「更懂生物」:论文最重要的启发不是参数量本身,而是证明了低表达/零表达基因和结构化先验知识,对单细胞 foundation model 来说都非常关键。
从应用角度看,scLong 展示出的潜力相当清晰。
首先,在基因扰动与功能研究中,它可以帮助研究者更快预测敲除、过表达、组合扰动可能带来的转录组变化,从而减少大量湿实验试错成本。
其次,在药物发现和精准医学中,它能够预测化学扰动和癌症药物反应,为候选药物筛选、联合用药设计和个体化治疗提供计算支持。
再次,在系统生物学层面,它还能辅助重建基因调控网络、理解细胞状态转换,并在多批次数据整合中提供更稳定的细胞表示。论文作者也指出,这样的模型有望进一步推动精准医疗、药物研发和细胞生物学研究。
更长远地看,scLong代表了一种很值得关注的方向:单细胞基础模型不应只是把Transformer搬到生物数据上,而应该同时拥抱「全局上下文」和「领域知识」。
当模型既能「读完整本基因之书」,又能理解每个基因在生物学中的位置,它才更有可能真正成为生命科学里的通用智能工具。
参考资料:
https://www.nature.com/articles/s41467-026-69102-y
文章来自于“新智元”,作者 “LRST”。


【开源免费】DeepBI是一款AI原生的数据分析平台。DeepBI充分利用大语言模型的能力来探索、查询、可视化和共享来自任何数据源的数据。用户可以使用DeepBI洞察数据并做出数据驱动的决策。
项目地址:https://github.com/DeepInsight-AI/DeepBI?tab=readme-ov-file
本地安装:https://www.deepbi.com/
【开源免费】airda(Air Data Agent)是面向数据分析的AI智能体,能够理解数据开发和数据分析需求、根据用户需要让数据可视化。
项目地址:https://github.com/hitsz-ids/airda
【开源免费】graphrag是微软推出的RAG项目,与传统的通过 RAG 方法使用向量相似性作为搜索技术不同,GraphRAG是使用知识图谱在推理复杂信息时大幅提高问答性能。
项目地址:https://github.com/microsoft/graphrag
【开源免费】Dify是最早一批实现RAG,Agent,模型管理等一站式AI开发的工具平台,并且项目方一直持续维护。其中在任务编排方面相对领先对手,可以帮助研发实现像字节扣子那样的功能。
项目地址:https://github.com/langgenius/dify
【开源免费】RAGFlow是和Dify类似的开源项目,该项目在大文件解析方面做的更出色,拓展编排方面相对弱一些。
项目地址:https://github.com/infiniflow/ragflow/tree/main
【开源免费】phidata是一个可以实现将数据转化成向量存储,并通过AI实现RAG功能的项目
项目地址:https://github.com/phidatahq/phidata
【开源免费】TaskingAI 是一个提供RAG,Agent,大模型管理等AI项目开发的工具平台,比LangChain更强大的中间件AI平台工具。
项目地址:https://github.com/TaskingAI/TaskingAI
【开源免费】XTuner 是一个高效、灵活、全能的轻量化大模型微调工具库。它帮助开发者提供一个简单易用的平台,可以对大语言模型(LLM)和多模态图文模型(VLM)进行预训练和轻量级微调。XTuner 支持多种微调算法,如 QLoRA、LoRA 和全量参数微调。
项目地址:https://github.com/InternLM/xtuner
